Superspreaders: Humans pass twice as many viruses to animals as we catch from them
The findings challenge our understanding of zoonotic diseases and highlight our integral role in the ecosystem's viral exchange.
The dreadful COVID pandemic brought zoonotic spillovers to the limelight. The leading theory is that SARS-CoV-2, the virus that causes COVID-19, mutated in bats and then jumped to other wild animals sold alive for food in the Wuhan markets, where they ultimately infected humans. The rest is, as they say, history.
This shouldn’t have come as a surprise as we’ve had to deal with many zoonotic diseases before, from Ebola to avian and swine influenzas. But it’s easy to forget that microbes do not make the cross-species leap in just one direction. It’s always a two-way street — and this dynamic can be astonishing.
Researchers at the University College London (UCL) have now investigated viral dynamics in unprecedented detail, revealing an eye-opening fact: humans pass twice as many viruses to domesticated and wild animals as we catch from them.
“Anthroponotics (human-to-animal diseases) are far more common than we thought with humans acting more as a source than a sink in the flow of viral exchange between host species,” Professor Francois Balloux of UCL and co-author of the new study told ZME Science.
The two-way street of viral infection
Anthroponosis, or the jump of viruses from humans to other animals, is increasingly being detected. Examples of such “reverse zoonotic” events include epidemics of the 2009 influenza A (H1N1) pandemic virus in pigs and epidemics of SARS-CoV-2 in minks and white-tailed deer. Pet owners may be more aware of this dynamic as it is common for dogs, cats, and even ferrets to show flu-like symptoms after close contact with infected humans.
Just how important of a viral source humans are was never evidently clear. And, of course, there’s a bias in this kind of research to focus on animal-to-human transmission. For their study, the researchers set about the herculean task of analyzing nearly 12 million viral genomes. These were used to build phylogenetic trees to help them find patterns of previous host jumps.
Imagine a scenario where a virus is found in both bats and humans, and the viral strains from these two hosts differ by only a few mutations. This similarity suggests a recent transmission event. But the critical question is: who infected whom? That’s what the phylogenetic trees are for.
Viral family trees
These phylogenetic trees are essentially family trees for viruses. By mapping out the evolutionary relationships between different viral strains, it’s possible to find their common ancestors. Such trees also allow scientists to infer the direction of a viral host jump.
“If we find well-supported clades of strains all from host species A embedded within a host species B clade, we can be pretty sure that the virus jumped from host B to A,” said Balloux.
This was no trivial task. The volume of data itself is immense. To make things even more challenging, the ecological diversity of viruses was much richer than previously classified using conventional methods. To overcome this problem, the researchers built their own phylogenetic tree that included 32 viral families based on genetic relatedness alone.
Another frustrating hiccup was the poor metadata at the researchers’ disposal. Many viral genomes were submitted to libraries missing vital information such as when, where, and which host the virus was sampled from.
“In fact, 45% of sequences were missing host information and 37% missing the date of sample collection. Additionally, we tend to focus on the viruses that infects humans, with 93% of all viral sequences being associated with humans. This massive sampling bias poses a great challenge to existing analytical approaches, and we spent a great deal of time ensuring that our results were not a product of it,” Cedric Tan, a Ph.D. student at UCL’s Genetics Institute and Francis Crick Institute, and lead author of the study, told ZME Science.
Humans as major viral contributors
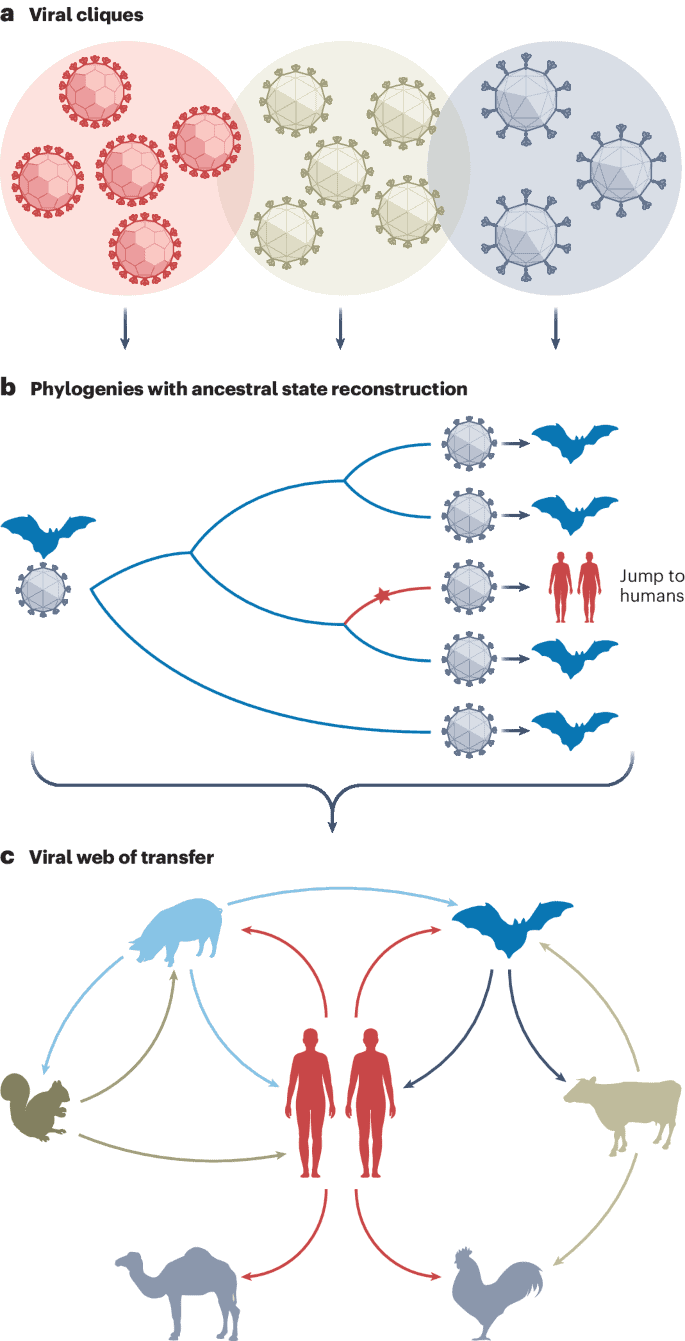
Through this meticulous reconstruction of viruses’ evolutionary histories and examination of genetic mutations acquired during host jumps, the researchers challenged the notion of humans as mere endpoints for viral diseases. Their most intriguing revelation is that human-to-animal transmission events are roughly twice as common as the reverse. Animal-to-animal host jumps that did not involve humans were even more common.
This once again underscores the fact that humans are never outside nature — although our hubris may lead us to think otherwise. Instead, we are integral parts of a complex ecosystem, endlessly exchanging pathogens with other species.
The study also explored the genetic underpinnings of these host jumps. They found that viruses often undergo significant mutations as they adapt to new hosts. Interestingly, viruses with a broad range of animal hosts showed fewer signs of such adaptive mutations. It would seem that there is an inherent versatility in infecting diverse species.
“Another interesting result is that viruses with a broad host range require less evolutionary changes to adapt to a novel host. This likely explains the well-known pattern that the viruses of most concern as possible agents for epidemics and pandemics tend to be generalists that can infect multiple hosts,” said Balloux.
Understudied but critical to consider
The findings have far-reaching implications, not only for understanding viral evolution but also for public health, conservation, and food security.
Viruses jumping from humans to animals may pose significant threats to wildlife conservation, potentially endangering species and disrupting ecosystems. For now, it’s unclear how much of a threat viruses jumping from humans are to other animals since this relationship is very understudied.
“The best-studied example is human influenza A H1N1 (part of the seasonal flu) which regularly causes epidemics in livestock and sometimes wildlife. Other known cases of reverse zoonoses have been described for a series of human viruses, including hepatitis E, rotavirus, herpesviruses, and adenoviruses. One example is the outbreaks of human metapneumovirus and human respirovirus 3 in wild chimpanzees (endangered species) in Uganda, 2016-2017, causing several deaths in the Chimpanzee community. This is likely only the tip of the iceberg as disease outbreaks in wild animals are only very rarely picked up,” said Balloux.
“One of the first steps to better understand the impacts of these host jumps is to bolster genomic surveillance efforts in domestic and wild animal species, which would help us to assess the diversity of viruses infecting animals, the frequency of host jumps, and the risk of emerging or re-emerging infectious diseases in these animals,” Tan added.
Zoonosis and disease risk
Reverse zoonosis could also make outbreaks among people more likely. Entirely new viruses can emerge via mutation or an exchange of genetic material among different viruses infecting a host at the same time. This latter process, known as reassortment, is particularly risky.
A prime example is the flow of viruses of humans from pigs, which act as mixing vessels that can then transmit new mutated strains back to humans. In 2009, the H1N1 flu virus killed between 151,700 and 575,400 people globally in its first year. But this virus, which jumped from animals to humans, comprised gene segments from four different origins: human, bird, the North American pig, and the Eurasian pig.
The degree to which reverse zoonosis increases the risk of pandemics or major outbreaks more broadly remains unclear. The majority of emerging zoonotic diseases have originated in wildlife, not livestock or pets. However, the interactions between humans and other species are incredibly complex, which warrants much more genomic surveillance than we currently undertake.
The findings appeared in the journal Nature Ecology & Evolution.









