Despite our differences, human beings share 99.9% of the genome. In other words, we all differ by a mere 0.1% of genes, which triggers the difference in the way we appear, grow, and develop.
Over 80% of rare diseases are caused by genetic mutations in that miniscule difference, and it’s estimated that such undiagnosed diseases affect about 8% of our population. Detecting such diseases is challenging, but researchers are working on new promising techniques.
Potential forms of diagnosis for rare and undiagnosed diseases include:
- Next Generation Sequencing (NGS), which refers to all large-scale DNA sequencing methods that allows for mapping the entire genome (whole genome sequencing);
- whole exome sequencing — focusing on just the exons within all known genes
- target gene panel (or only exons of selected genes).
To understand whole exome sequencing (WES), we need dive into the world of our genetic makeup.
Four letters
The nucleus of every cell in the human body consists of 23 pairs of chromosomes, which makes 46 chromosomes in every cell. These chromosomes are in turn made of double stranded DNA.
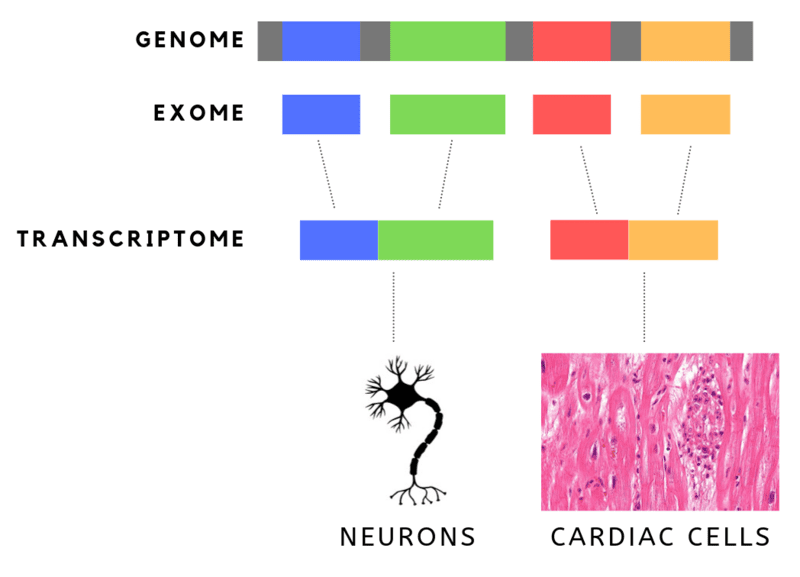
DNA is made up of genes that are built on nucleotides. The human genome consists of 20,000 genes and 3 billion nucleotides or “letters.” The’ letters’ are organic molecules, namely- Guanine (G), Thymine (T), Adenine (A) and Cytosine (C). G, T, A and C are arranged in specific sequences in our genes, subsequences that translate into proteins.
But not all 3 billion nucleotides translate into proteins. In fact, only a small percentage (about 1.5%) of these nucleotides, are actually translated into proteins. These are “EXpressed regiONS”, or exons.
This has led to the herald of Whole Exome Sequencing, or WES. While the cost of sequencing the entire genome is still out of reach, the cost of sequencing just the exons (aka the functional part of the genome) is low enough that it has been used to find genetic abnormalities leading to rare diseases. It is also much easier to sift through this data.
The complementary, “INTragenic regiON” or introns in genes are not represented or translated in proteins.
| Whole Exome Sequencing | Whole Genome Sequencing |
| Sequencing only the “coding DNA” or 30 million lettersLess intensive analytically and has lower storage requirementsLess expensive($1000 commercially)High sequencing depth of protein coding regions Ability to detect certain types of alterations may be limited Includes newly characterized and novel genes | Sequencing “all” DNA , introns and exons- 3 billion lettersMore variants to analyze and more storage requirements Much more expensive ($20,000 commercially)Extensive and uniform coverage of genome at a lower sequencing depth, both protein coding and non coding regions of genome Can detect more types of alterations than exome sequencing Includes newly characterized and novel genes Can detect up to 10-15% more diagnoses than WES |
The National Organization for Rare Disorders (NORD) at NCSU hosted Dr. Vandana Shashi, a pediatric genetics specialist at Duke University on April 22. Shashi she served as the co-chair of NIH’s Rare and Undiagnosed disease network and shared her perspectives on Exome and Genome Sequencing.
Given the nature of the method, some alterations that are not reliably detected by WES include- deep intronic non-coding region defects, pseudogenes and repeat regions. As WGS becomes cheaper and more accessible, Dr. Shashi sees this method eclipsinging WES.
“I do see WGS becoming cheaper and more accessible in the future, this will eventually eclipse WES,”Dr Shashi said, “WGS is a lot better at capturing copy number variants, i.e, deletions and duplications that are larger than 50 base pairs.”

In Whole Exome Sequencing, there are three steps DNA is prepared, sequenced and processed.
Step 1- DNA Library Preparation
- Shear DNA – First, genomic DNA is sheared into random short fragments of about 300 base pairs.
- Blunting- When an enzyme is used to chop the DNA into small parts, it leaves ends of uneven length on the double strand, depending on which strand is larger and which is shorter, the base pairs are either removed, or the missing base pairs are filled in by an enzyme producing “blunt” ends of equal length.
- A-tailing- The blunted ends are then modified by adding a single adenine (A) nucleotide that forms an overhanging “A-tail”.
- Add adapters- The sample is flanked by ligate adaptors to allow sequencing.
- Enrich library for Exome capture- Sequences that correspond to exons are captured by hybridization to DNA or RNA baits and then pulled down by coated magnetic beads. The selected fragments create a library enriched with exomes.
Step 2- DNA Sequencing

Exome capture is followed by amplification of the sample and massive parallel sequencing. Massively parallel second generation sequencing (aka next generation sequencing) generates billions of base pairs of data. Barcodes to allow sample indexing, can be introduced at this step.
“You attach DNA to the flow cells and amplify. Basically, you are doing a number of simultaneous PCR reactions (Multiplex PCR reactions). Then you read the sequence and you get a lot of fragments,” Dr Shashi said,”these fragments come in short reads of 100-115 base pairs long,”
Step 3- DNA Analysis
The next step is DNA Alignment and Variant calling.

“These fragmented base pairs from the previous step are overlapped with one another and they are compared against a reference genome,” Dr. Shashi said.
A reference genome here is a so-called ‘normal genome’ or a representative example of the set of genes in one idealized individual of the human species.
Bioinformatics tools are then used in DNA analysis, they usually use one of these three file formats-
| FASTQ | BAM | VCF |
| It offers full sequencing of data and a corresponding quality score. Each sequence filtering gets entered as a 4 line format. Very large file formats, requires a lot of storage space. | Binary Alignment Map, facilitates alignment of FASTQ to a reference genome Very large file formats, requires a lot of storage space | Variant Call FormatStandardized text file for representing Single Nucleotide Polymorphisms (SNP), Insertions and Deletions in the genome (INDEL) and corresponding variationsMost commonly used format |

Courtesy Twist Biosciences
Dr Shashi used this method to diagnose a 20 month old with Brown–Vialetto–Van Laere Syndrome 2 (BVVLS2) (Shashi et. al) , they used high-dose riboflavin therapy or large doses of Vitamin B2 to stabilize the degressing neurological condition of the child.
Whole exome Sequencing is shaping up to be the most exciting advance in the world of genetics and could possibly be a much larger stepping stone in the world of undiagnosed and rare disorders. Stay tuned as we keep up with this evolving bio technology.







{kind=link}